We have heard alot about STAT-2 trial.
There are two big questions and one was not even mentioned.
(1). The trial finishes and then what happens?
ProfG rehearsed some of the arguments yesterday and if/(when) pharma treatments for progressive MS become licenced in the meantime e.g. siponimod, it creates complexity. They have a few years to think of a response but it should have been thought through already.
Maybe they have?
Maybe the MHRA (UK regulators) have given advice?.
It said in a paper (Telegraph) "The trial is scheduled to last six years, but because statins are already known to be safe and widely used" (yes but not at the dose in the trial the usual maximum is 40mg/day not 80mg/day, which some people may not tolerate well), "the treatment would quickly pass through regulators if it was deemed to be successful" (Has this been agreed?, Will the regulators readily accept a trial without a dose-response? The need for the high-dose, over the standard dose has not been made..or has it? The MHRA wouldn't care about the cost required of doing a low-dose arm. Cost is not their concern. Is it unimportant? If there are alternatives available surely they could require that one to do two trials like they ask industry to do? Anyway as there are thirty centres this trial will give neuros the experience and hence confidence to use off-label as it is hard to see anyone applying for or holding a licence (the ministers were quite clear there is no need for an off-label licencing bill and it is undesirable for governments to be asked to hold licenses and so doctors can prescribe off-label if they want...Will there be a change in Government between then and 6 years in the future?) and could be good news for pwMS in UK, but will a post-Brexit EMA be accomodating and what would the take up be outside of the UK?).
(2). However what was not mentioned in the media is
What is the working mechanism that statins are targeting?
We are asking over 1000 thousand people to be involved onto such a trial and this has not been addressed except:
How do statins work?
There are two big questions and one was not even mentioned.
(1). The trial finishes and then what happens?
ProfG rehearsed some of the arguments yesterday and if/(when) pharma treatments for progressive MS become licenced in the meantime e.g. siponimod, it creates complexity. They have a few years to think of a response but it should have been thought through already.
Maybe they have?
Maybe the MHRA (UK regulators) have given advice?.
It said in a paper (Telegraph) "The trial is scheduled to last six years, but because statins are already known to be safe and widely used" (yes but not at the dose in the trial the usual maximum is 40mg/day not 80mg/day, which some people may not tolerate well), "the treatment would quickly pass through regulators if it was deemed to be successful" (Has this been agreed?, Will the regulators readily accept a trial without a dose-response? The need for the high-dose, over the standard dose has not been made..or has it? The MHRA wouldn't care about the cost required of doing a low-dose arm. Cost is not their concern. Is it unimportant? If there are alternatives available surely they could require that one to do two trials like they ask industry to do? Anyway as there are thirty centres this trial will give neuros the experience and hence confidence to use off-label as it is hard to see anyone applying for or holding a licence (the ministers were quite clear there is no need for an off-label licencing bill and it is undesirable for governments to be asked to hold licenses and so doctors can prescribe off-label if they want...Will there be a change in Government between then and 6 years in the future?) and could be good news for pwMS in UK, but will a post-Brexit EMA be accomodating and what would the take up be outside of the UK?).
(2). However what was not mentioned in the media is
What is the working mechanism that statins are targeting?
We are asking over 1000 thousand people to be involved onto such a trial and this has not been addressed except:
How do statins work?
We know the simple answer is.
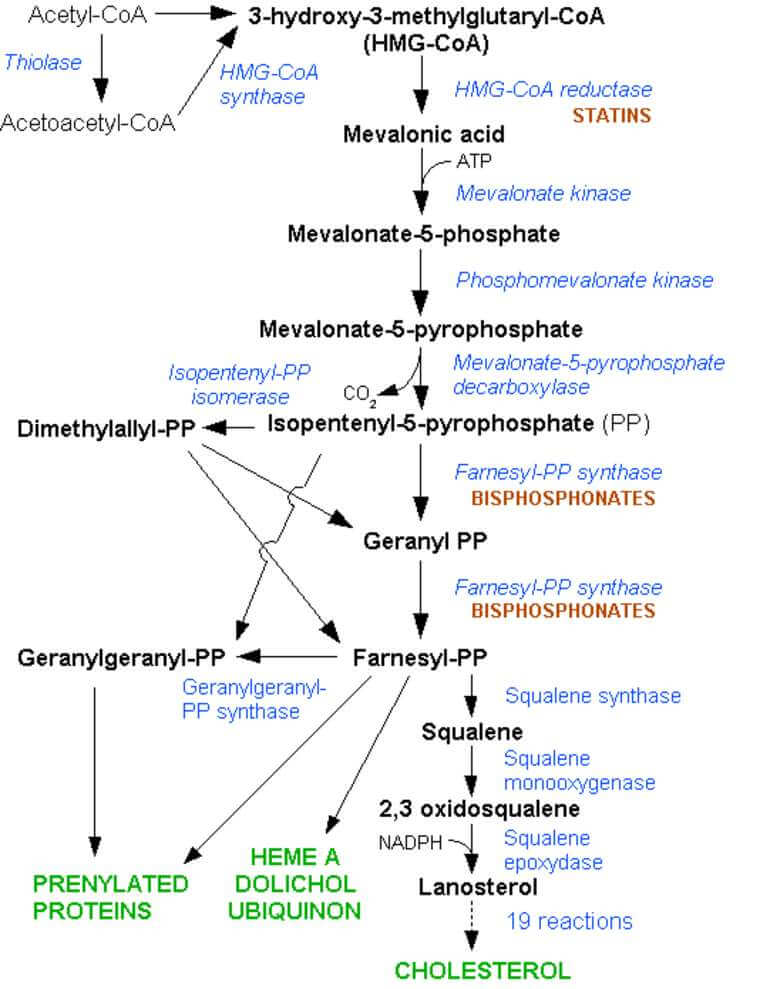
Statins are HMG-coA reductase Inhibitors and so block the production of cholesterol and any molecule upstream of this (see the diagram below)
However this is a cop-out answer not saying anything about what it is doing in MS.
However this is a cop-out answer not saying anything about what it is doing in MS.
What we want to know is.
How do statins work in MS?
This is not a trick question, but I wonder if you ask most of the neurologists in the 30 centres associated with this trial, will they have a sensible answer ready prepared and supplied by the trial co-ordinators, because if the answer to the question is "I don't know", is that OK?
The original suggestion was part based on our own work. It for started for us with a couple of blokes called John Greenwood (Blood brain barrier interest) and Pete Adamson (Rho signalling interest) at the UCL Institute of Ophthalmology, London
They were trying to work out how cells got into the brain from the blood. They had found that the lymphocytes signal to the endothelial cell in the blood vessel wall and that the endothelial cell would then remodel its interal skeleton to allow a cell to push through the blood vessel wall. (Yes cells go through the cell not through the junctions of the cells).
This was known to involve a molecule called Rho. Rho affects the skeletal changes by a process involving prenylation (also known of lipidation) where isophenyl precursors get prenyl chemical groups added to then to activate the remodelling of the skeleton made of a protein called actin. Simply put carbon containing molecules are added to make a long chain.
This was known to involve a molecule called Rho. Rho affects the skeletal changes by a process involving prenylation (also known of lipidation) where isophenyl precursors get prenyl chemical groups added to then to activate the remodelling of the skeleton made of a protein called actin. Simply put carbon containing molecules are added to make a long chain.
What Prof John Greenwood realised was that the isophenyl precursors for this event were made via the cholersterol pathway. So block the isoprenyl precursors and you stop cell migration across the blood brain barrier.
We had done it with Geranyl and Farnesyl PP inhibitors, so why not a statin further up the chain. This blocks HMG-Co A which is limiting in the cholesterol synthesis pathway.
We had done it with Geranyl and Farnesyl PP inhibitors, so why not a statin further up the chain. This blocks HMG-Co A which is limiting in the cholesterol synthesis pathway.
So MD2 and I did the experiment and low and behold statins (we used lovastatin) inhibited EAE.
I heard that Professor Larry Steinman (a Science neuro at Stanford University, USA), who has more Nature papers than you can shake a stick at, also had similar animal data with a molecule called Atorvastatin.
However, they were saying it worked by a cytokine switch to make Th1 (pro-inflammatory cells) into Th2 (anti-inflammatory cells (see education section if you want this explained).
This was sort of boring because everything that stopped EAE at the time, did the Th1 to Th2 switch. (We thought this not a sensible approach, as Th2 cells would be bad in MS too :-().
However, we had the same in vivo data in animals, but a completely different mechanism of action. So I suggested we submit the two papers together to Nature, hoping that Larry's pull may get our paper in, and the controversy would get his paper in. So we submitted "back to back" papers.
I heard that Professor Larry Steinman (a Science neuro at Stanford University, USA), who has more Nature papers than you can shake a stick at, also had similar animal data with a molecule called Atorvastatin.
However, they were saying it worked by a cytokine switch to make Th1 (pro-inflammatory cells) into Th2 (anti-inflammatory cells (see education section if you want this explained).
This was sort of boring because everything that stopped EAE at the time, did the Th1 to Th2 switch. (We thought this not a sensible approach, as Th2 cells would be bad in MS too :-().
However, we had the same in vivo data in animals, but a completely different mechanism of action. So I suggested we submit the two papers together to Nature, hoping that Larry's pull may get our paper in, and the controversy would get his paper in. So we submitted "back to back" papers.
Guess what happened:-). How wrong was I?
Greenwood J, Walters CE, Pryce G, Kanuga N, Beraud E, Baker D, Adamson P. Lovastatin inhibits brain endothelial cell Rho-mediated lymphocyte migration and attenuates experimental autoimmune encephalomyelitis. FASEB J. 2003;17(8):905-7.
One look at the experiments (see D below) showed us that the Th2 switch , which was a flavour "mechanism of the month" at that time. Bet it would be T regulatory cells now) was probably nonsense. (However, other people also claimed this mechanism. They discovered the trick to find Th2 cytokines). The Blue line is the control showing relapses occurring. You can see in red that disease was inhibited by the statin until about day 47 when all animals got disease. However, we stopped treating on day 45, the drug is out of the system in 48h and you got disease. Nice work MD2! In our mind a Th2 cell would not have reverted (to a Th1) in 48h or ever.In (A) drug was stopped on day 25 and disease resumed. (B) Histology showing lesions did not occur (C) Treatment at onset didn't make much difference so it is not treatng the attack. You use steroids for this.

If you want to read the science, you can read.
Nath N, Giri S, Prasad R, Singh AK, Singh I. Potential targets of 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor for multiple sclerosis therapy. J Immunol. 2004; 172(2):1273-86.
Greenwood J, Steinman L, Zamvil SS Statin therapy and autoimmune disease: from protein prenylation to immunomodulation. Nat Rev Immunol. 2006;6:358-370.
There were some other groups doing stuff and also it was suggested to block microglial activity
So before you can say "translational science", some clinicians were on the case and trials were being done.
Those Americans don't hang around.
Those Americans don't hang around.
Vollmer T, Key L, Durkalski V, Tyor W, Corboy J, Markovic-Plese S, Preiningerova J, Rizzo M, Singh I. Oral simvastatin treatment in relapsing-remitting multiple sclerosis. Lancet. 2004;363:1607-8.
So our idea translated into humans as lesion numbers were dropped, but it wasn't great. Beta interferon would be beating this But a 80mg dose of Simvastatin (which was a more potent anti-T cell proliferative agent than others) was plucked out of the air and used.
Another trial was suggested to be positive on lesions, but then the rot set in.
A small study with interferon beta suggested worsening and interest began to fail in RRMS, although worsening was not found in other studies.
Sorensen PS, Lycke J, Erälinna JP, Edland A, Wu X, Frederiksen JL, Oturai A, Malmeström C, Stenager E, Sellebjerg F, Sondergaard HB; SIMCOMBIN study investigators. Simvastatin as add-on therapy to interferon β-1a for relapsing-remitting multiple sclerosis (SIMCOMBIN study): a placebo-controlled randomised phase 4 trial. Lancet Neurol. 2011;10(8):691-701.
but not all the science was positive
Miron VE, Zehntner SP, Kuhlmann T, Ludwin SK, Owens T, Kennedy TE, Bedell BJ, Antel JP. Statin therapy inhibits remyelination in the central nervous system. Am J Pathol. 2009;174(5):1880-90
However, the UK tortoise eventually got there but I guess the lack of novelty in RRMS prompted a study in Progressive MS and MS-STAT was born.
(I never asked what whas the original logic of doing this study).
It was the first small molecule to have an effect on progressive disease.
(I never asked what whas the original logic of doing this study).
It was the first small molecule to have an effect on progressive disease.
Chataway J, Schuerer N, Alsanousi A, Chan D, MacManus D, Hunter K, Anderson V, Bangham CR, Clegg S, Nielsen C, Fox NC, Wilkie D, Nicholas JM, Calder VL, Greenwood J, Frost C, Nicholas R. Effect of high-dose simvastatin on brain atrophy and disability in secondary progressive multiple sclerosis (MS-STAT): a randomised, placebo-controlled, phase 2 trial. Lancet. 2014;383:2213-21.
The mean annualised atrophy rate was significantly lower in patients in the simvastatin group (0·288% per year [SD 0·521]) than in those in the placebo group (0·584% per year [0·498]). The adjusted difference in atrophy rate between groups was -0·254% per year (95% CI -0·422 to -0·087; p=0·003); a 43% reduction in annualised rate. Simvastatin was well tolerated, with no differences between the placebo and simvastatin groups in proportions of participants who had serious adverse events (14 [20%] vs nine [13%]).
INTERPRETATION: High-dose simvastatin reduced the annualised rate of whole-brain atrophy compared with placebo, and was well tolerated and safe. These results support the advancement of this treatment to phase 3 testing.
So the data says it may slow progression, it does not say it halts progression.
As we have said many times before you probably need this in addition to a DMT. However, this could be problematic as there is now the baggage of it worsening beta interferon treated MS in a Cochrane Review.
Why secondary Progressive MS?
Maybe it was genius of John Greenwood and Jeremy Chataway to pick progressive MS, because we have shown that the major dysregulated pathway in progressive EAE, is the cholesterol pathway.
Sevastou I, Pryce G, Baker D, Selwood DL. Characterisation of Transcriptional Changes in the Spinal Cord of the Progressive Experimental Autoimmune Encephalomyelitis Biozzi ABH Mouse Model by RNA Sequencing.PLoS One. 2016;11(6):e0157754.
It is also the major pathway that is dysregulated in Alzheimers disease, which is known to be neurodegenerative. I suspect it will be within these pathways that statins are going to be active in progressive MS.
Chang TY, Yamauchi Y, Hasan M, Chang CC. Cellular Cholesterol Homeostasis in Alzheimer's Disease. J Lipid Res. 2017 Mar 15. pii: jlr.R075630.
Chang TY, Yamauchi Y, Hasan M, Chang CC. Cellular Cholesterol Homeostasis in Alzheimer's Disease. J Lipid Res. 2017 Mar 15. pii: jlr.R075630.
Indeed Statins may be protective in Alzheimers and it is good to see the data that Simvastatin and Atorvastatin are better than pravastatin. This is because pravastatin does not get into the CNS very well and this would therefore suggest that the benefit in Alzheimers is due to activity in the CNS, rather than affecting a co-morbidity that would not require CNS-penetrance of the statin.
Zissimopoulos JM, Barthold D, Brinton RD, Joyce G. Sex and Race Differences in the Association Between Statin Use and the Incidence of Alzheimer Disease. JAMA Neurol. 2017 Feb 1;74(2):225-232. doi: 10.1001/jamaneurol.2016.3783.
IMPORTANCE:To our knowledge, no effective treatments exist for Alzheimer disease, and new molecules are years away. However, several drugs prescribed for other conditions have been associated with reducing its risk.
OBJECTIVE: To analyze the association between statin exposure and Alzheimer disease incidence among Medicare beneficiaries.
DESIGN, SETTING, AND PARTICIPANTS: We examined the medical and pharmacy claims of a 20% sample of Medicare beneficiaries from 2006 to 2013 and compared rates of Alzheimer disease diagnosis for 399 979 statin users 65 years of age or older with high or low exposure to statins and with drug molecules for black, Hispanic, and non-Hispanic white people, and men and women of Asian, Native American, or unkown race/ethnicity who are referred to as "other."
MAIN OUTCOMES AND MEASURES: The main outcome was incident diagnosis of Alzheimer disease based on the International Classification of Diseases, Ninth Revision, Clinical Modification. We used Cox proportional hazard models to analyze the association between statin exposure and Alzheimer disease diagnosis for different sexes, races and ethnicities, and statin molecules.
RESULTS:The 399 979 study participants included 7794 (1.95%) black men, 24 484 (6.12%) black women, 11 200 (2.80%) Hispanic men, 21 458 (5.36%) Hispanic women, 115 059 (28.77%) white men, and 195 181 (48.80%) white women. High exposure to statins was associated with a lower risk of Alzheimer disease diagnosis for women (hazard ratio [HR], 0.85; 95% CI, 0.82-0.89; P<.001) and men (HR, 0.88; 95% CI, 0.83-0.93; P<.001). Simvastatin was associated with lower Alzheimer disease risk for white women (HR, 0.86; 95% CI, 0.81-0.92; P<.001), white men (HR, 0.90; 95% CI, 0.82-0.99; P=.02), Hispanic women (HR, 0.82; 95% CI, 0.68-0.99; P=.04), Hispanic men (HR, 0.67; 95% CI, 0.50-0.91; P=.01), and black women (HR, 0.78; 95% CI, 0.66-0.93; P=.005).
Atorvastatin was associated with a reduced risk of incident Alzheimer disease diagnosis for white women (HR, 0.84, 95% CI, 0.78-0.89), black women (HR, 0.81, 95% CI, 0.67-0.98), and Hispanic men (HR, 0.61, 95% CI, 0.42-0.89) and women (HR, 0.76, 95% CI, 0.60-0.97).
Pravastatin were associated with reduced Alzheimer disease risk for white women only (HR, 0.82, 95% CI, 0.70-0.95 and HR, 0.81, 95% CI, 0.67-0.98, respectively). High statin exposure was not associated with a statistically significant lower Alzheimer disease risk among black men.
CONCLUSIONS AND RELEVANCE: The reduction in Alzheimer disease risk varied across statin molecules, sex, and race/ethnicity.
Pravastatin were associated with reduced Alzheimer disease risk for white women only (HR, 0.82, 95% CI, 0.70-0.95 and HR, 0.81, 95% CI, 0.67-0.98, respectively). High statin exposure was not associated with a statistically significant lower Alzheimer disease risk among black men.
CONCLUSIONS AND RELEVANCE: The reduction in Alzheimer disease risk varied across statin molecules, sex, and race/ethnicity.
Geifman N, Brinton RD, Kennedy RE, Schneider LS, Butte AJ.Evidence for benefit of statins to modify cognitive decline and risk in Alzheimer's disease. Alzheimers Res Ther. 2017 Feb 17;9(1):10.
This was notable in people homozygous for ApoE (variant4,) (ApoE4 is am Alzheimer risk factor gene and in some MS studies too. Although most people with MS have ApoE3). ApoE transports cholesterol.
The question is How does it Work?
Here are some possibilities
1. Simvastatin does nothing in SPMS. The phase II results
were a fluke (I doubt it, but it is possible).
2. Simvastatin does nothing directly to SPMS, but affects the co-morbidities (heart function, fat metabolism) that predispose to worse
course/health. This is why you need a plausible mechanism to get your research teeth into.
3. Inhibits lesion formation by blocking blood brain barrier dysfunction. Although this may be of some benefit in MS, the failures of this
approach in SPMS, one would suggest this is not of major impact.
4. Promotes vascular perfusion. I know what you are thinking, but it has been suggested to me, by one of the team.
4. Promotes vascular perfusion. I know what you are thinking, but it has been suggested to me, by one of the team.
4. Inhibits T cell activation/proliferation and promotes the
production of T cell inhibitory cytokines. Based on the many failures of this
approach in RRMS (?) and SPMS, one would think this is not of major impact.
5. Inhibits B cell activation/proliferation. Blocking B cell activity
has some influence on progressive MS. Simvastatin gets into the CNS, unlike
pravastatin and can affect B cell function.
6. Block microglial migration and function, such as nitric oxide release and free-radical formation. This is important to progression and other neurodegenerative diseases.
7. Block of Astrocyte activity, such as via inhibition of Nuclear Factor-kappaB transcription factor
7. Block of Astrocyte activity, such as via inhibition of Nuclear Factor-kappaB transcription factor
8. Influence neurological compensation by neurogenesis and synapse formation,
although dendritic spine formation is RhoA-dependent and this could be blocked.
9. Influence on myelination. Myelination is all about lipid/fatty sheath formation. Although there are reports of improvements in myelination, there are more reporting negative influences on myelination. One would think this is not of major impact.
10. Block the production of oxidised cholesterols
(oxysterols), which are cytotoxic, to oligodendrocytes possibly due to potassium overload, and nerves. This is a major damaging mechanism proposed in Alzheimers disease. Oxysterols are increased in the brains of people with MS and is is CNS occuring event. This mechanism can target and disrupt mitochondria (the power house of the cell. This is where I would suggest an activity could be occurring.
The CNS does not rely largely on cholesterol from systemic circulation due to limited metabolic turnover during adulthood and the brain’s inherent capacity to synthesise its own cholesterol. Unlike cholesterol in plasma which has a half-life of only a few days, brain cholesterol has been associated with a half-life of from 6 months to 5 years. Thus, chronic statin therapy may be required before significant effects on CNS cholesterol are seen.
So are they going to re-baseline their scans after a few months to allow the drug to work.
24(S)-Hydroxycholesterol escapes the brain and may be used as a monitor of activity. Is it being measured I don't know.
So are they going to re-baseline their scans after a few months to allow the drug to work.
24(S)-Hydroxycholesterol escapes the brain and may be used as a monitor of activity. Is it being measured I don't know.
It is not my Job to do the reading to explain the logic of trials, so maybe one of the many Neuros attached to the trial like ProfG can come up with a few more, after all, it is they who have to explain why it is a good idea to participate in this study.
However I have looked on the MS Society website who co-funded the trial and tehre is nothing mentioned there except it is a cholesterol inhibiting drug.
However I have looked on the MS Society website who co-funded the trial and tehre is nothing mentioned there except it is a cholesterol inhibiting drug.
If you fit the selection criteria and you have secondary progressive MS, you may wish to participate.
You can get info here.
https://www.mssociety.org.uk/ms-stat2-info
You can get info here.
https://www.mssociety.org.uk/ms-stat2-info
So whilst we give the Neuros (Sir Jeremy) a pat on the back, remember Prof Greenwood, was the scientist that got this all going in the first place and we played our part in this bit of MS history.